
If your company develops software for a medical device, the international standard IEC 62304 is the global benchmark for a safe and compliant software development lifecycle. Regulators around the world, including the FDA and European authorities, recognize it as the accepted state of the art for medical software.
Achieving compliance requires a disciplined and documented approach to software development, maintenance, and risk management. This cannot be done with informal or ad hoc processes. It requires a robust IEC 62304 QMS (Quality Management System) that provides the structure, procedures, and controls needed to meet the standard’s demanding requirements. This guide will serve as a high level checklist for what your QMS must cover to make sure your medical software is compliant, safe, and ready for regulatory submission.
Table of Contents
What Is IEC 62304 and Why It Matters
This section will define the standard and explain its part in medical device software development. It establishes the “why” before diving into the “how.”
IEC 62304 is the international standard that defines the lifecycle requirements for the development of medical device software. Its full title is “Medical device software – Software life cycle processes.” The standard provides a detailed framework of processes, activities, and tasks that are necessary to guarantee the safe design, development, and maintenance of medical software. It matters because it is a harmonized standard. This means it is officially recognized by regulatory bodies globally as the expected methodology for developing safe medical software.
Compliance with IEC 62304 is the clearest way to demonstrate to regulators that you have followed a rigorous and repeatable process. The standard is risk based, which means the amount of effort and documentation required is scaled according to the potential harm the software could cause if it fails. The standard defines three Software Safety Classes (A, B, and C), from lowest risk to highest risk, and the level of rigor required for a Class C device is higher than for a Class A device. An IEC 62304 QMS provides the necessary framework to manage this entire lifecycle according to your software’s specific class. It is not just a standard for your developers; it is a standard that your entire quality and regulatory organization must understand and support. Any modern QMS in medical device development that involves software must be built to accommodate its requirements.
A compliant QMS is the key to proving to auditors and regulators that your software was developed in a state of control from its initial concept all the way through to its final retirement.
The Role of a Quality Management System (QMS)
An IEC 62304 QMS for life sciences is the operational framework that allows you to execute the requirements of the standard in a controlled, repeatable, and auditable manner. The standard itself tells you what you need to do, for example, create a software development plan, manage risk, and test your software. Your QMS, on the other hand, defines how your specific organization will do it.
Your QMS will contain the specific Standard Operating Procedures (SOPs), work instructions, and templates that your teams will use to create all the required documentation. It is the system that manages your software requirements specifications, your risk management file, your architectural design documents, your testing protocols, and all your software release records. A modern electronic QMS (eQMS) is all-important for managing this level of complexity. An eQMS can enforce your development workflows, manage the critical links between requirements, code, tests, and risks, and provide a complete, auditable design history file (DHF) for regulators at the push of a button. Building an IEC 62304 QMS is a foundational step for any company serious about developing medical software, including innovative products like SaMD (Software as a Medical Device).
Mapping IEC 62304 to ISO 13485 Requirements
There is a very close and important relationship between IEC 62304 and ISO 13485. While IEC 62304 focuses specifically on the software lifecycle, ISO 13485 defines the requirements for the overall Quality Management System for a medical device manufacturer. The two standards are designed to work together in harmony. A key part of building an IEC 62304 QMS is integrating its specific software requirements into your broader ISO 13485 certified QMS.
ISO 13485 requires you to have documented processes for design and development, risk management, and supplier control, but it is not specific about how to manage the unique challenges of software. IEC 62304 provides that specific, detailed guidance for the software components of your device.
Here is how they typically map:
Design and Development Controls (ISO 13485, Clause 7.3)
The entire IEC 62304 software development process, from planning and requirements analysis through to final release, is considered the detailed execution of this clause for your software. The deliverables required by IEC 62304, such as your software requirements specification and architectural design, directly fulfill the design input and output requirements of ISO 13485.
Risk Management (ISO 13485, Clause 7.1)
IEC 62304 requires a specific software Risk Management process that must be part of your overall device risk management process, which is governed by the ISO 14971 standard. The software hazards you identify feed into the main device risk file.
Purchasing and Supplier Controls (ISO 13485, Clause 7.4)
If you use any third party software, including open source libraries or off the shelf software (SOUP), IEC 62304 has specific and demanding requirements for managing and documenting those components. This directly aligns with the supplier and purchasing control requirements of ISO 13485. The importance of ISO 13485 in medical device development is that it provides the quality system umbrella under which specific standards like IEC 62304 operate.
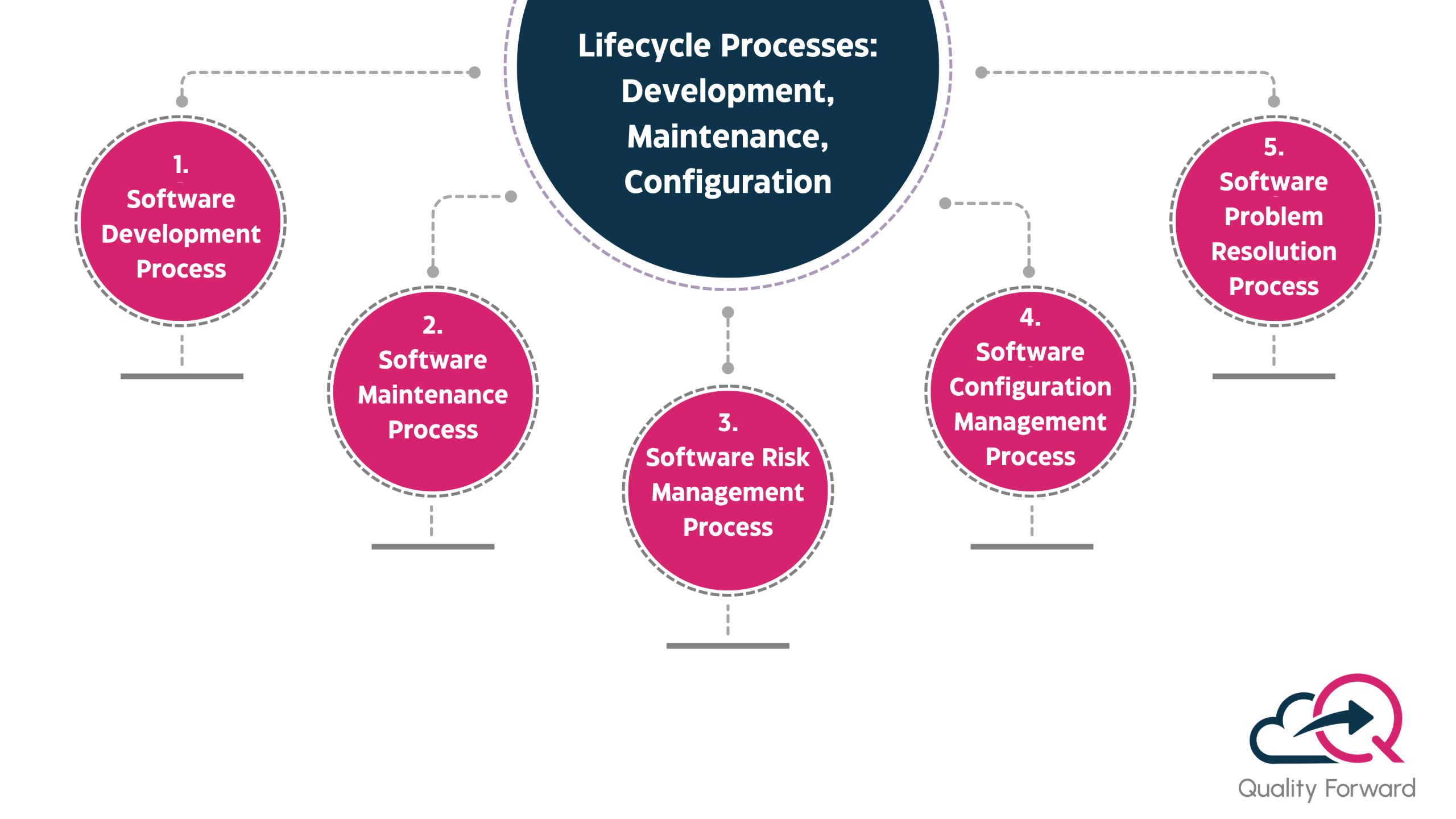
Lifecycle Processes: Development, Maintenance, Configuration
The core of IEC 62304 is its definition of the software lifecycle processes. Your IEC 62304 QMS must have clear and detailed procedures and controls for each of these key areas.
- Software Development Process: This is the main part of the standard. It requires a structured process that includes software development planning, software requirements analysis, architectural design, detailed software design, unit implementation and verification, software integration and integration testing, and final software system testing. Each of these stages requires specific documented outputs that become part of your auditable design history file.
- Software Maintenance Process: Software is never truly “done.” The standard requires a documented process for managing your software after it has been released to the market. This includes a system for monitoring user feedback, evaluating bug reports, and making changes in a controlled and documented manner. Any change, no matter how small, must go through a risk based process to ensure it doesn’t introduce a new or unexpected hazard.
- Software Risk Management Process: This process is critical and must be active throughout the entire lifecycle. You must have a systematic way of identifying potential software hazards, estimating the associated risks, and implementing control measures to reduce those risks to an acceptable level. This Risk Management process must be documented in a software risk management file and must be integrated with your overall device risk file according to ISO 14971.
- Software Configuration Management Process: You need a robust system to control and uniquely identify all the different components that make up your software. This includes your source code, your build scripts, your configuration files, and all your third party libraries. This process guarantees that you can reliably and repeatedly build and release your software and that you have a complete, auditable history of all changes made to the codebase.
- Software Problem Resolution Process: This process defines how you will track, evaluate, and resolve all problems and bugs found, both during development and after release. Each problem report must be assessed for its potential impact on safety, and the resolution must be properly tested and verified. A strong IEC 62304 QMS will often manage this within the CAPA or non conformance module of the eQMS.
You can learn more about the official standard on the IEC 62304:2006 page.
Common IEC 62304 Pitfalls and How to Avoid Them
Achieving and upholding compliance with IEC 62304 can be challenging. There are several common problems that companies, especially those new to medical device development, frequently run into.
Poor or Incomplete Documentation
The standard is heavy on documentation. A common failure is treating this documentation as an afterthought to be completed at the end of the project. To avoid this, you must “document as you go,” making it a natural part of the development process. An eQMS can help by providing templates and enforcing documentation steps within the workflow.
Inadequate Risk Management
Many teams fail to perform a thorough software risk analysis, or they don’t properly link their risk management activities to their design choices and testing protocols. To avoid this, your risk file should be a living document that is reviewed and updated with every code change.
Weak SOUP Management
Using third party software, or Software of Unknown Provenance (SOUP), without properly evaluating its risks and documenting its use is a major compliance gap. Your IEC 62304 QMS must have a clear procedure for identifying, evaluating, and documenting every piece of third party code used in your device.
Lack of Traceability
A very frequent audit finding is the inability to show a clear, documented link, or traceability, from a software requirement to its design, to the code that implements it, to the test that verifies it, and to the risk controls associated with it. An eQMS with built in traceability features is the best way to avoid this.
Treating Maintenance as an Afterthought
The maintenance process requires the same level of rigor and documentation as the initial development process. Cutting corners on a “minor” bug fix is a common mistake that can introduce serious new risks into a previously safe product.
An IEC 62304 QMS is your best defense against these problems by providing a structured framework that guides your team. All of these areas fall under the umbrella of modern QMS standards.
Conclusion
Compliance with IEC 62304 is a huge requirement for any company developing medical device software. It is the accepted global standard for demonstrating that your software is safe and was developed in a controlled, repeatable manner. Building a strong IEC 62304 QMS that is fully integrated with your overall ISO 13485 quality system is the only way to achieve this efficiently and effectively. The standard is detailed and demanding, but it provides a clear roadmap for success.
At Quality Forward, our eQMS is designed to support the rigorous documentation, traceability, and risk management requirements of standards like IEC 62304. We provide a validated, centralized platform for managing your entire software lifecycle, from requirements to release and maintenance. If you are developing a medical software product and need a QMS that can keep up with the demands of agile development and regulatory compliance, contact us to see how we can simplify your compliance journey.
FAQs: IEC 62304 and Quality Management Systems (QMS)
A quality management system (QMS) supports IEC 62304 compliance by providing structured processes for documentation, risk management, software change control, and traceability. A digital QMS like Quality Forward enables automated workflows, version control, and audit-ready records aligned with regulatory expectations.
Yes. While the FDA and EU MDR do not mandate IEC 62304 by name, they expect software development practices that align with its principles. IEC 62304 is recognized by the FDA and harmonized under the EU MDR for software as a medical device (SaMD) and embedded software in medical devices.
Key documentation includes a software development plan, risk management files, verification and validation plans, software release notes, and traceability matrices. A compliant QMS ensures all these records are stored, versioned, and accessible for audits and regulatory submissionsץ
IEC 62304 defines three software safety classes:
Class A – No injury or damage to health is possible
Class B – Non-serious injury is possible
Class C – Death or serious injury is possible
The QMS should reflect the rigor required for each class during development and post-market processes.
An electronic QMS (eQMS) streamlines documentation by managing version control, automated approvals, and audit trails. With a system like Quality Forward, development teams can link requirements, risk assessments, testing, and change control records for full IEC 62304 traceability.
Preparation involves ensuring all software lifecycle activities are documented and traceable. This includes having up-to-date SOPs, complete risk assessments, documented testing, and software release procedures. Using a validated QMS ensures audit readiness and reduces the risk of findings
Yes, but additional documentation and retrospective risk assessments may be required. A compliant QMS helps organize legacy evidence, apply risk controls, and document updates to meet IEC 62304 expectations – even for previously developed software.


