
For any quality assurance professional in a regulated manufacturing environment, few phrases cause as much anxiety as “The FDA is here.” An FDA audit is a rigorous, in-depth examination of your facility, processes, and records, and the outcome can have a massive impact on your business. But it doesn’t have to be a source of stress. With the right preparation, a culture of quality, and robust systems, an audit can be a smooth process that confirms the strength of your operations. The key is readiness. This guide provides a comprehensive FDA audit checklist to help your QA team prepare, understand the process, and demonstrate a state of constant compliance, making sure you are always ready for an inspection.
Table of Contents
What Is an FDA Audit and Why It Matters in Manufacturing QA
An FDA audit, formally known as an inspection, is the agency’s primary method for checking that regulated companies are complying with the law. For manufacturers, this means verifying adherence to Current Good Manufacturing Practices (cGMP) and other relevant regulations. The goal of the inspection is to protect public health by making sure that drugs and medical devices are produced in a safe, controlled, and consistent manner. An FDA audit is not a punitive measure by default, it’s a verification activity. However, the findings can lead to serious consequences if major deficiencies are found. For a QA team, the audit is the ultimate test of your systems. It’s a direct evaluation of your document control, training programs, deviation handling, and overall ability to produce a safe and effective product. A successful outcome is a testament to your hard work, while a poor outcome can signal deep-rooted problems in your quality management system.
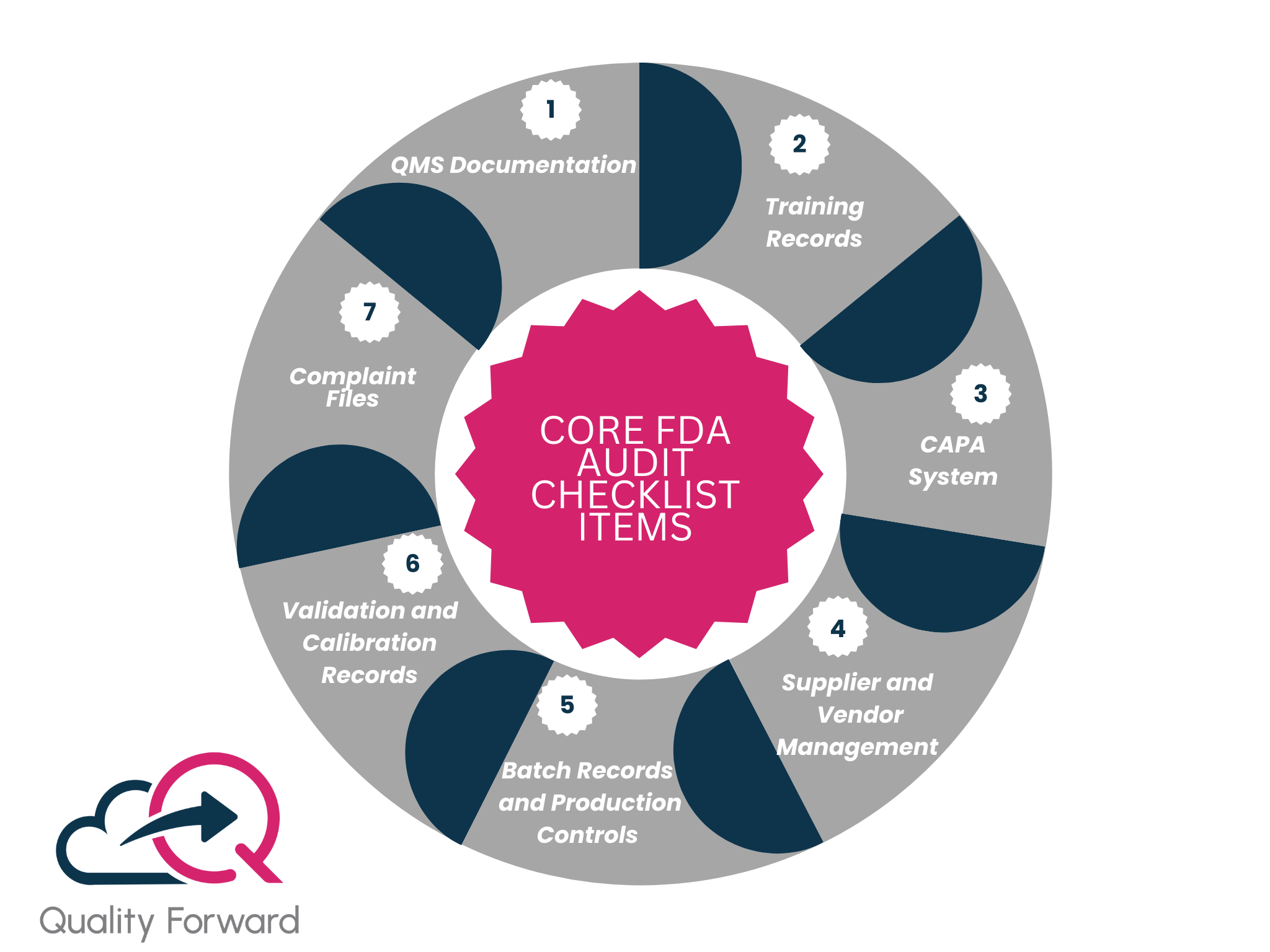
Core FDA Audit Checklist Items for Manufacturing Teams
While every FDA audit is unique, inspectors typically focus on a core set of systems and records that provide a comprehensive view of your quality operation. Being “audit-ready” means having these areas in perfect order at all times. Use this FDA audit checklist as a guide for your internal reviews and preparation activities to make sure you have all your bases covered. An inspector should be able to ask for any of these items and receive a prompt, accurate, and complete response.
Quality Management System (QMS) Documentation
This is the starting point. Be ready to present your Quality Manual, organizational charts, and key SOPs. The inspector will want to see a well-defined, documented system that is understood and followed by your team. This includes your procedures for document control and record retention.
Training Records
You must be able to prove that all employees are thoroughly trained for their specific roles. Inspectors will randomly select employees and ask to see their complete training files, including their initial training, ongoing refreshers, and any role-specific qualifications. Gaps in training records are a common and easily avoidable finding.
Corrective and Preventive Action (CAPA) System
Your CAPA system is a direct measure of how you handle problems and improve your processes. Be prepared to show your logs for deviations, non-conformances, and OOS results. The inspector will review specific CAPA records to see if your root cause investigations are thorough, your corrective actions are effective, and you are verifying that the problem has been solved.
Supplier and Vendor Management
You are responsible for the quality of the materials you get from your suppliers. Your FDA audit checklist must include your supplier qualification files, which should contain your audits, quality agreements, and performance monitoring records for all critical suppliers.
Batch Records and Production Controls
The inspector will want to review a selection of completed batch records to verify that they are complete, accurate, and show that the product was manufactured according to the validated process. They will check for signatures, data entries, and the documentation of any in-process deviations.
Validation and Calibration Records
You must have proof that your equipment, processes, and facilities are fit for their intended use. Be ready to provide your validation master plan, as well as specific validation reports (IQ/OQ/PQ) and routine calibration and maintenance records for key pieces of equipment.
Complaint Files
How you handle customer complaints is a critical indicator of your post-market surveillance. The inspector will review your complaint handling procedure and specific complaint files to check they are investigated properly, trended, and linked to your CAPA system if necessary.
Preparing Your QA Processes for an FDA Audit
Beyond having your documents in order, true audit readiness involves preparing your people and processes. A frantic, last-minute attempt is a recipe for mistakes. Instead, you should cultivate a state of constant readiness so that an unannounced FDA audit is business as usual.
- Conduct Regular Mock Audits: The best way to prepare for an audit is to practice. Use your FDA audit checklist to conduct rigorous internal audits or hire a third party to simulate the pressure and scrutiny of a real inspection. This helps identify weaknesses in a low-stakes environment.
- Establish Clear Audit Roles: Don’t wait until the inspector is in the lobby to decide who does what. Designate a “back room” team to retrieve documents, a “front room” team (including your subject matter experts) to answer questions, and a “scribe” to log all requests and documents provided.
- Prepare Your Subject Matter Experts (SMEs): Your SMEs should be ready to speak confidently and accurately about their areas of expertise. Coach them to answer only the question that is asked, to be honest if they don’t know an answer, and to never guess or speculate.
- Review Past Audit Findings: Look at your own internal audit findings, as well as any past regulatory inspections. Confirm that all previous corrective actions have been fully implemented and verified as effective. Inspectors will almost always check on past promises.
- Organize Your Records for Quick Retrieval: Whether your records are paper-based or electronic, you need to be able to pull them quickly. A long delay in finding a requested document can give the impression of disorganization or, worse, that you are hiding something. This is where modern audit management in manufacturing software can provide a huge advantage.
Understanding FDA Form 483
If an FDA investigator observes conditions that, in their judgment, may constitute violations of the Food, Drug, and Cosmetic Act, they will issue an FDA Form 483 at the conclusion of the inspection. It’s so important for your team to understand what this form is, and what it isn’t. An FDA Form 483 is not a final determination of non-compliance. It is a list of observations that the inspector believes are out of line with regulations. It’s an opportunity for you to respond and correct the issues.
Receiving a 483 is serious, and recent data shows that regulatory scrutiny is increasing. As the FDA ramped up inspections following the pandemic, the number of observations surged. According to official FDA data, the number of Form 483s issued to drug establishments in FY2022 increased by about 116% over the previous year. The trend was even more pronounced for medical device companies, which saw an increase of almost 200% in the same period. This data makes it clear that audit readiness is more important than ever.
So, what are inspectors actually finding? The data points to consistent failures in foundational quality systems. For drug manufacturers, the most frequently cited issue between FY2018 and FY2022 was related to 21 CFR 211.22(d), meaning the Quality Control team failed to establish or follow its own written procedures. For medical device companies, the top citation was for 21 CFR 820.100(a), pointing to inadequate procedures for Corrective and Preventive Action (CAPA). These stats show that mastering the basics is how to avoid common audit findings.
If you do receive an FDA Form 483, your response is so important. You must respond in writing, typically within 15 business days, addressing each observation with a clear plan for corrective action. Your response should be thorough, convincing, and demonstrate that you take the findings seriously. A weak or slow response can escalate the situation, potentially leading to a formal warning letter. To better understand the types of issues that lead to regulatory action, QA teams can review the warning letters published by FDA on their public database, which provides real world examples of compliance failures.
Best Practices for Passing FDA Audits
Passing an FDA audit consistently comes down to a few core principles. First is transparency. Never try to hide a problem or mislead an inspector. It’s far better to own a known issue and present your plan to fix it than to be caught in a lie, which destroys all credibility. Second is organization. A smooth, well-run audit where documents are retrieved quickly and SMEs are prepared gives the inspector confidence in your systems. This is where simplifying audit management with digital tools can transform the experience. Third, demonstrate a clear and robust “quality culture.” This means showing that quality is not just the QA department’s job, but that everyone from production to the executive level is invested in it. This is evident in clean facilities, well-maintained equipment, and employees who can speak knowledgeably about their role in supporting product quality. An FDA audit is as much an audit of your culture as it is of your paperwork.
Why QMS Is Critical During FDA Audits
A modern, electronic Quality Management System (eQMS) is arguably the single most important tool for audit readiness. A centralized eQMS in manufacturing takes every item on the FDA audit checklist and makes it easier to manage and present. Document control is automated, making sure only the current approved versions are in use. Training records are linked to employee roles and automatically tracked. CAPAs, deviations, and complaints are managed in a structured workflow with a complete, unchangeable audit trail. When an inspector asks for a record, it can be found with a few clicks instead of a frantic search through filing cabinets. This level of organization and data integrity is what inspectors expect to see in a modern facility. Simply put, the best QMS is one that transitions your audit preparation from a panicked, manual event into a state of continuous, calm readiness, making any FDA audit a much more manageable process.
Conclusion
An FDA Audit should be viewed as an opportunity to demonstrate the strength and effectiveness of your quality systems. The key to success lies in continuous preparation. By using an FDA audit checklist to guide your internal efforts and by building your quality operations on a foundation of robust, modern systems, you can face any inspection with confidence.
At Quality Forward, we specialize in helping regulated companies prepare for the highest levels of scrutiny. Our expertise in quality assurance and the validation of systems like eQMS guarantees that your digital tools are not only efficient but fully compliant and audit-ready. If you’re looking to strengthen your audit preparedness and build a world-class quality system, contact us to see how we can help.
Frequently Asked Questions (FAQs): FDA Audit Checklist
An FDA audit checklist serves as a structured tool to evaluate whether a regulated organization meets key requirements for regulatory compliance, data integrity, process control, and documentation. It enables systematic review of quality-system elements and inspection readiness.
Key focus areas include documentation and record control, employee training and competency, deviation and CAPA (Corrective and Preventive Action) management, equipment and facility controls, process validation, change control, supplier oversight, and internal audit and management review processes.
Organizations should maintain an up-to-date audit checklist aligned with applicable regulations and standards, perform regular internal audits, confirm procedures are being followed, verify documentation traceability, ensure training is complete, and test retrieval of essential records to demonstrate inspection readiness.
A QMS provides the structure behind many checklist elements; including SOPs, audit trails, change control, CAPA, training, and management review. A well-implemented QMS helps maintain consistency, traceability, and evidence that processes meet FDA expectations.
The audit checklist should be reviewed and updated whenever there are changes to regulations, company processes, systems, or products, or after significant audit findings. At a minimum, annual review is recommended to ensure ongoing compliance and readiness for inspection.
Common findings include missing or outdated SOPs, incomplete training documentation, CAPAs lacking verification of effectiveness, poor change-control documentation, incomplete supplier qualification records, and challenges retrieving documents during inspections.
Mock inspections simulate an FDA inspection to evaluate how well an organization can respond, retrieve records, and demonstrate control of its processes. They help identify weaknesses early so corrective actions can be implemented before an actual inspection.
Using an electronic QMS (eQMS) system provides centralized control of documents, electronic audit trails, real-time data access, version control, training tracking, and integration with other systems such as CAPA and change control. These capabilities improve visibility, speed up document retrieval, and support overall inspection readiness.


