
Imagine you’re managing a global Phase III clinical trial for a potentially life-saving new drug. You have 40 investigator sites spread across 15 countries, a lead contract research organization (CRO) in the US, a central lab in Europe, and a data management vendor in Asia. Each day, you’re dealing with different time zones, languages, and local regulations, all while thousands of critical data points are being generated.
The future of your product, the return on hundreds of millions of dollars in investment, and most importantly, the safety of every patient, all depend on the integrity of that data. In an environment this complex and high-stakes, how do you maintain control? How do you make sure everyone, everywhere, is following the same protocol? The answer is a robust QMS in clinical trials. This is the operational framework that provides the structure, controls, and oversight needed to manage the immense complexity of a modern trial and support its success.
Table of Contents
What is a QMS in the Context of Clinical Trials?
A Clinical Quality Management System is a formal system of documented processes, procedures, and responsibilities designed to guarantee that a clinical trial consistently meets its quality objectives while adhering to all regulatory requirements. While a QMS in a manufacturing plant is built around Good Manufacturing Practice (GMP) to guarantee product consistency, the world of a QMS in clinical trials is built on the principles of Good Clinical Practice (GCP). GCP, as defined by the International Council for Harmonisation (ICH E6 R2/R3), is the international ethical and scientific quality standard for how trials involving human subjects should be designed, conducted, recorded, and reported.
The primary goals of a Clinical Quality Management System are to protect the rights, safety, and well-being of trial participants and to confirm the clinical data is credible, accurate, and reliable. It is a comprehensive system that touches every single aspect of a trial, defining how the protocol is developed and amended, how sites are selected and initiated, how documents are controlled in the Trial Master File (TMF), how staff are trained, how deviations are managed, and how you maintain rigorous oversight of every single vendor and partner involved in the study.
Why a Generic QMS Isn’t Enough for Clinical Trials
Some organizations, especially those with a strong manufacturing background, are tempted to simply adapt their existing QMS for their clinical operations. This approach is almost always a mistake. Clinical trials present a unique set of challenges and risks that demand a purpose-built system. The controlled, predictable environment of a factory is a world away from the dynamic, decentralized nature of a global clinical trial.
A QMS for clinical trials must be designed to handle the specific documentation, workflows, and risks of the clinical environment. For example, managing the Trial Master File (TMF) is a monumental task with no equivalent in manufacturing. The TMF is a living collection of thousands of documents from dozens of sources that must be complete, accurate, and inspection-ready at all times, a challenge that requires specialized eTMF capabilities.
The entire operational model of a clinical trial is different. You are managing the performance of dozens of independent investigator sites, each with its own staff, facilities, and procedures. You are also outsourcing critical functions to CROs and other vendors. You are ultimately responsible for the quality of their work, and a generic QMS lacks the specific tools needed to manage these complex third-party relationships, oversee their performance, and handle the flow of data and documents between multiple organizations. The focus on patient-centric risks, such as the integrity of the informed consent process and the timely reporting of adverse events, requires specific, dedicated workflows that simply don’t exist in a standard manufacturing QMS.
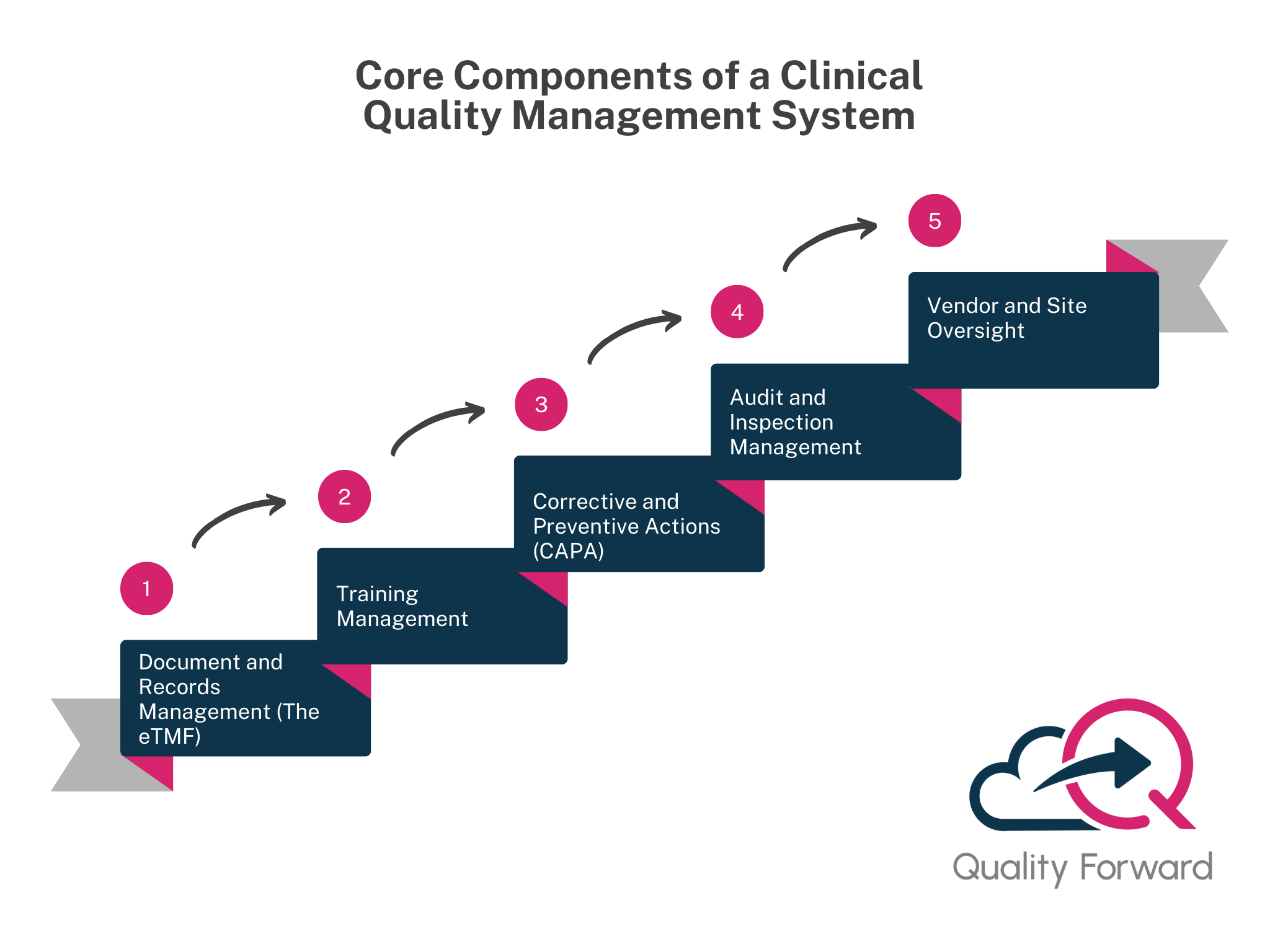
Core Components of a Clinical Quality Management System
A strong QMS for clinical trials is built on several key pillars. These components work together to create a state of control and proactive quality management, guaranteeing your trial is always audit-ready.
Document and Records Management (The eTMF)
This is the heart of any clinical QMS. It’s about the active, real-time management of the electronic Trial Master File (eTMF). The system must control every document, the protocol and its amendments, the investigator’s brochure, informed consent form templates, regulatory approvals, and ethics committee correspondence. It must make sure that only the single, current, approved version is accessible to all sites globally. A bulletproof, unchangeable audit trail for every action (view, edit, approval, signature) is a non-negotiable requirement for both Good Manufacturing Practice (GMP) and GCP. A best-in-class system will align with industry standards like the DIA TMF Reference Model, which provides a standardized structure for the TMF, making audits and inspections infinitely smoother.
Training Management
Trial’s success depends on hundreds of people across multiple organizations performing their roles correctly. The QMS must manage and document training for this incredibly diverse group, your internal team (CRAs, data managers), the principal investigators and nurses at each clinical site, and the project team at your CRO. The system needs to link specific training requirements to roles and protocol versions, automatically flag when retraining is needed (e.g. after a protocol amendment), and be able to produce a complete, auditable training record for any individual in a matter of seconds. Without this, proving personnel are qualified is a nightmare of chasing paper..
Corrective and Preventive Actions (CAPA)
When things inevitably go wrong in a trial, a site consistently deviates from the protocol, a data discrepancy trend is found, or a patient misses a critical visit, you need a structured, documented process to deal with it. A clinical CAPA process is unique, it’s designed to investigate these issues, perform a thorough root cause analysis, and implement effective Corrective and Preventive Actions (CAPAs) to prevent them from happening again. For example, if multiple sites are making the same data entry error, a good CAPA process would identify the confusing section of the electronic case report form (eCRF) as the root cause and the corrective action would be to clarify it and retrain all sites. This system is a direct measure of your organization’s commitment to learning and improving.
Audit and Inspection Management
Your QMS must serve as the command center for your entire audit universe. This includes planning and executing your risk-based internal audit program to find your own problems before regulators do. It covers scheduling and conducting audits of your clinical sites (to ensure GCP compliance) and your key vendors (to verify they are meeting their quality agreements). And, crucially, it manages regulatory inspections. When an inspector arrives, the QMS is where you log their requests, track the provision of documents, and manage any subsequent findings and responses.
Vendor and Site Oversight
You can outsource the work, but you can never outsource the ultimate responsibility for quality and patient safety. Your QMS must have a module for qualifying your vendors (CROs, central labs, e-diaries), documenting your due diligence, and managing formal quality agreements that clearly define expectations. It should also track vendor performance over time, including their handling of deviations, data queries, and CAPAs. This gives you the documented oversight you need to confirm every partner in your trial is meeting your quality standards and maintaining GxP Compliance.
The Critical Role of Data Integrity and FDA 21 CFR Part 11
As clinical trials have become almost entirely dependent on electronic systems, the integrity of the data those systems hold is everything. This is precisely why regulators established FDA 21 CFR Part 11. This regulation sets the binding requirements for guaranteeing that electronic records and electronic signatures are just as trustworthy, reliable, and legally equivalent as paper records with a handwritten signature.
For any QMS in Clinical Trials that uses electronic systems, from the eTMF and EDC systems to the interactive voice response systems (IVRS) used for patient randomization, compliance with FDA 21 CFR Part 11 mandates specific technical and procedural controls. This includes secure, computer-generated, time-stamped audit trails that must track the creation, modification, or deletion of any electronic record. The audit trail must record the “who, what, when, and why” of any change. The rule also requires strict, role-based access controls to make sure that only authorized individuals can access the system and perform their specific job functions. It requires the use of secure, unique electronic signatures that are backed by a clear attestation. This regulation is the absolute base of trust for all of the electronic data that your trial generates and upon which your submission will depend.
Key Benefits of a Robust QMS in Clinical Trials
Trying to manage a global clinical trial using a patchwork of disconnected tools like spreadsheets, email, and various shared drives is not just inefficient, it is a profound compliance risk. A centralized, modern Clinical Quality Management System provides the integrated control needed for success.
- Improved Data Quality and Integrity: By enforcing standardized processes, providing secure audit trails, and reducing manual data handling, a QMS dramatically reduces the risk of human error and confirms that the data you collect is reliable, defensible, and of the highest possible quality.
- Faster Study Execution and Startup: A well-implemented QMS streamlines the administrative workflows that cause delays, helping to get trials up and running more quickly. This speed is critical in an industry where the cost of a one day delay in bringing a drug to market can be in the millions. Modern QMS platforms often incorporate disciplined quality methodologies like Six Sigma, and the impact is significant. One study demonstrated that applying such techniques can lead to a 70% reduction in cycle-time for data-heavy processes like Case Report Form data entry.
- Simplified Audits and Inspections: When an auditor asks for the training record for a specific nurse at a site in another country, a centralized QMS allows you to retrieve that document in seconds. This ability to respond quickly and confidently demonstrates a state of control and makes the entire inspection process far less stressful and more successful.
- Better Oversight of CROs and Vendors: A QMS provides a single, unified platform to manage all vendor qualification documents, track key performance indicators from your quality agreement, and guarantee that all your external partners are adhering to the same high quality standards.
- Reduced Overall Compliance Risk: A QMS provides a structured framework to proactively identify, manage, and mitigate the myriad of risks inherent in clinical research. This makes serious, costly compliance failures far less likely to occur.
Conclusion
In clinical development, where patient safety and data integrity are all-important, a powerful QMS in clinical trials is a necessity. It provides the framework for navigating the complex web of global regulations and for managing the operational complexities of modern research. By choosing an eQMS tailored for life sciences, you move beyond simply collecting data and begin actively managing quality across your entire trial. To learn more about the specific rules governing trial conduct and reporting, stakeholders can review the Official legal requirements and related resources. The best QMS will always be one that brings control, clarity, and confidence to your clinical operations.
At Quality Forward, we understand the unique pressures of GxP environments and the specific demands of Good Clinical Practice. We provide expert software validation and quality assurance services to make sure that the critical systems you rely on for your clinical trials are fully compliant, reliable, and ready for the deepest regulatory scrutiny. If you are implementing a new QMS or need to guarantee your existing systems are validated correctly, contact us to see how our expertise can aid in your trial’s success.
Frequently Asked Questions (FAQs): QMS for Clinical Trials
A QMS in the clinical-trial context is a structured framework of policies, standard operating procedures (SOPs), work instructions and records designed to assure participant safety, data integrity and regulatory compliance across the full lifecycle of a clinical trial.
Implementing a QMS helps ensure consistent execution of trial processes, supports audit-readiness, reduces repetitive quality issues and strengthens regulatory posture.
Relevant regulatory frameworks include ICH E6 (R2) (Good Clinical Practice), ISO 14155:2020 for medical-device investigations, and region-specific regulations such as Regulation (EU) No 536/2014. Additionally, in the US context, regulations such as 21 CFR Part 50, 56, 312 or 812 may apply depending on the trial type.
Foundational aspects include leadership commitment, quality-focused culture, defined governance and alignment to organizational objectives.
Core elements typically include document and record control, risk management, training and competency, issue and deviation management, knowledge management, audit readiness, and management review.
A clinical QMS centers on trial conduct, subject safety, data integrity and regulatory compliance for human research. By contrast, a manufacturing QMS emphasizes product quality, production processes and post-market surveillance.
A robust QMS should be embedded across all phases, from pre-trial planning and protocol design, through study conduct and monitoring, to study close-out and post-trial review.
Early integration of risk assessment and SOP development in startup phases helps avoid downstream quality issues and audit findings.
Key steps include performing a gap assessment (against frameworks such as those by TransCelerate Biopharma), defining quality policies and objectives, mapping core processes and controls, implementing SOPs and training, and establishing metrics and management review cycles.
Also important is fostering an organizational culture of quality, maintaining documentation traceability and leveraging suitable electronic QMS (eQMS) tools when appropriate.
An eQMS offers digital workflows, audit trails, real-time document control, training tracking, CAPA management, and integration with other clinical systems (e.g., eTMF, CTMS).
Using an eQMS can reduce manual effort, enhance transparency and traceability, mitigate error-prone paper-based processes, and better support inspection readiness.


