
Working in life sciences means living with constant oversight. Whether you are developing new treatments, building medical devices, or running trials, there is always a standard to meet and a regulator to answer to. Everyone expects quality, and for good reason. But meeting those expectations across different countries and industries is not always straightforward, especially when we talk about QMS standards.
That is why QMS standards matter. They give your team a clear foundation to work from – a shared understanding of how processes should be documented, risks managed, and improvements made over time. These standards are not just about staying compliant. They help teams stay consistent, avoid mistakes, and keep products safe from start to finish.
In the sections that follow, we will walk through the major standards that matter most in life sciences. ISO, FDA, and EU guidelines, and explain where each one fits. We will also look at how digital tools can take some of the pressure off, especially when your team is managing multiple requirements across regions. Whether you are setting up a quality system for the first time or reviewing your existing setup, this guide will give you a clearer view of what to focus on and how to stay ready.
Table of Contents
What Are QMS Standards
Quality management standards are the rules and frameworks that help companies run quality systems with structure and reliability. They are not just for passing audits. They guide how teams document processes, manage risk, handle complaints, and improve over time.
For life sciences companies, these standards are not just best practices. They are expectations. Whether you are developing medicine, launching a new device, or running clinical trials, regulators expect your quality system to follow these guidelines and show that you are in control of your work.
Good quality management standards give you the tools to build that control into everyday operations. They help reduce errors, support accountability, and give your team a clear system to follow when things go wrong or need to improve.
Differences Between ISO, FDA and EU Standards
Not all QMS standards are the same. While they share similar goals, the specific requirements and focus areas can vary depending on the industry and region.
ISO Standards
These are internationally recognized and apply across many sectors. ISO 9001 is one of the most widely used standards and focuses on general quality principles. ISO 13485 builds on this but is more targeted to medical devices, with stronger emphasis on risk management and regulatory alignment.
FDA Standards
If your product is sold in the United States, the FDA has specific rules. 21 CFR Part 820 outlines how medical device manufacturers should manage their quality systems. It covers everything from design controls to production and post-market reporting. For electronic records, companies must follow FDA 21 CFR PART 11, which ensures digital systems are secure, traceable, and audit friendly.
EU Standards
In the European Union, EU Annex 11 sets the bar for computerized systems used in regulated environments. It works closely with GMP requirements and focuses on validation, system access, and data integrity. If you are using an eQMS in Europe, Annex 11 compliance is essential.
ISO AND FDA QMS Compliance
Many life sciences companies must meet both ISO and FDA QMS compliance. This means choosing tools and workflows that support multiple standards without creating confusion or duplicate effort.
QMS Standard: ISO 9001 Compliance
ISO 9001 is one of the most widely adopted QMS standards, offering a flexible framework for building quality into everyday business processes. It is designed to apply across industries but has proven especially valuable in life sciences where precision, traceability, and compliance are critical.
Clause 4 of the standard focuses on understanding the context of the organization, which means defining internal and external issues that could impact your quality objectives. For a pharmaceutical or medical device company, this might involve evaluating supply chain risk or shifting regulatory expectations.
Clause 6 introduces the concept of risk-based thinking, encouraging teams to identify potential issues before they occur and build controls that prevent them. This is not about adding more paperwork. It is about making better decisions with fewer surprises. For example, when a process deviation is logged, teams should be able to trace it back to a documented risk and review whether the controls in place were strong enough.
Clause 8 outlines operational planning and control. In life sciences, this includes process validation, documented procedures, and clear acceptance criteria. This clause helps teams stay focused on consistency and product safety by making sure every step is planned and monitored.
Meeting ISO 9001 QMS requirements helps life sciences teams create a structured way to monitor performance, handle non-conformances, and keep continuous improvement at the center of operations. When supported by digital tools like an eQMS, the standard becomes far more manageable, giving teams the ability to implement quality with less admin and more confidence.
ISO 9001 quality management is not just about checking boxes during audits. It builds habits that support collaboration, clarity, and readiness across departments. From supplier evaluations to internal reviews, it helps everyone stay on the same page and focused on delivering high-quality, compliant results with high QMS standard.
ISO 9001:2015 vs. ISO 9001:2026
The upcoming ISO 9001:2026 revision is set to build on the foundation of ISO 9001:2015 while introducing clearer expectations for digital maturity, sustainability, and operational resilience.
In life sciences, where documentation, traceability, and compliance are non-negotiable, this shift reinforces the need for modern quality systems that go beyond manual processes and isolated tools. The 2026 version is expected to emphasize topics such as data integrity, change control, and proactive risk mitigation, all areas where traditional QMS tools often fall short.
Quality teams preparing for these updates should evaluate how well their systems support connected workflows, audit readiness, and continuous improvement. A cloud-based eQMS with integrated modules for deviations, CAPAs, document control, and training provides the structure needed to adapt quickly and maintain compliance.
Aligning with ISO 9001:2026 compliance won’t just be about meeting a checklist. It will mean rethinking quality as a dynamic, integrated function that supports both regulatory compliance and long-term business resilience.
QMS Standard: ISO 13485 Compliance
ISO 13485 is the international standard specifically developed for quality management systems in the medical device industry. It builds upon ISO 9001 but includes more rigorous and detailed requirements focused on product safety, regulatory compliance, and risk management throughout the entire device lifecycle.
At its core, ISO 13485 emphasizes the need for a documented quality system that supports both design and manufacturing controls. Clause 4 of the standard outlines general requirements for a QMS in manufacturing, including maintaining documented procedures, controlling outsourced processes, and managing quality records. Clause 4.2 specifically highlights the need for a quality manual, documented procedures, and controlled records to demonstrate compliance and traceability.
Design and development controls, which are not mandatory in ISO 9001, are required under Clause 7.3 of ISO 13485. This clause mandates planning, input and output reviews, verification and validation activities, and documented changes. This ensures that every medical device is developed systematically and in compliance with regulatory expectations.
Risk management is a central theme in ISO 13485 and is referenced across multiple clauses, including Clause 7.1, which states that risk must be considered throughout product realization. This ties directly to ISO 14971, the standard for risk management of medical devices, reinforcing the need to identify, evaluate, and mitigate risks throughout the lifecycle of the product.
Control of suppliers and purchased products is another important aspect. Clause 7.4 requires manufacturers to evaluate and monitor suppliers to ensure that purchased products and services meet defined requirements. Supplier controls are essential in an industry where component quality directly impacts patient safety.
Clause 8 focuses on measurement, analysis, and improvement. It includes requirements for internal audits (Clause 8.2.4), corrective and preventive actions (Clause 8.5), and nonconformance management (Clause 8.3). A robust audit management system can help teams track findings, implement follow-ups, and close out issues while maintaining complete documentation.
Traceability is also a critical part of ISO 13485. Clause 7.5.3.2 requires that manufacturers establish systems for identifying the product throughout production and, in some cases, post-delivery. This is particularly important for implantable devices, where full traceability to individual patients may be required.
Implementing ISO 13485 often involves integrating a dedicated eQMS that can support the complexity of medical device development and production. Document control, training records, supplier qualifications, and risk assessments all need to be tightly managed. With the right system in place, teams can align daily operations with the standard’s expectations and be fully prepared for audits, inspections, and market submissions.
Whether you are launching a new Class II device or maintaining compliance for an existing product portfolio, ISO 13485 provides the structure and clarity needed to meet global regulatory demands and consistently deliver safe, effective medical devices.
QMS Standard: 21 CFR PART 820 Compliance
This regulation, often referred to as the Quality System Regulation, is issued by the United States Food and Drug Administration and applies specifically to medical device manufacturers. It outlines the minimum requirements for quality systems to ensure that medical devices are safe, effective, and consistently produced. Unlike voluntary quality management standards such as ISO 9001, compliance with 21 CFR Part 820 is mandatory for companies that want to market their devices in the United States.
The regulation covers every part of the product lifecycle and is a QMS standard. That includes how products are designed, how manufacturing is controlled, how complaints are handled, and how records are maintained. Each process must be clearly documented, followed as written, and regularly reviewed. The goal is to ensure that companies can demonstrate full traceability and control over what they make and how they make it.
There are several key areas that manufacturers need to pay close attention to:
- Design Controls
Companies must have a formal process for planning, reviewing, verifying, validating, and documenting product design. These controls help ensure that the final product meets user needs and intended use. - Documented Procedures
Every process affecting quality must be governed by clear procedures. These procedures need to be accessible, kept up to date, and followed by all relevant personnel. - Production and Process Controls
Manufacturers are expected to monitor and control how devices are made. This includes validating critical processes, training operators, and maintaining equipment to avoid variability. - Corrective and Preventive Actions (CAPA)
When issues are found, they must be investigated thoroughly. The root cause should be identified, and appropriate actions must be taken to prevent recurrence. - Complaint Handling
All complaints must be documented, evaluated, and investigated when necessary. This includes determining whether the issue is reportable under FDA regulations. - Recordkeeping and Traceability
Every action and decision related to product quality must be recorded. Records must be easy to retrieve and stored securely for the required retention period.
Failure to comply with 21 CFR Part 820 can have serious consequences. Companies may face warning letters, import restrictions, fines, or forced recalls. That is why many teams rely on a digital system to manage these requirements. A modern eQMS can help track documentation, automate training, link audit findings to CAPAs, and ensure everything is documented in real time.
QMS Standard: GxP and GMP
GxP refers to a family of quality regulations that apply to life sciences, where safety, efficacy, and data integrity are critical. These include Good Manufacturing Practice (GMP), Good Laboratory Practice (GLP), and Good Clinical Practice (GCP). Each one focuses on a different phase of the product lifecycle, but they all share a core principle: quality must be built into every step of your operations.
Good Manufacturing Practice (GMP) outlines the minimum requirements manufacturers must meet to ensure products are consistently high in quality and safe for use. This includes strict guidelines on facility hygiene, equipment calibration, validated manufacturing processes, environmental monitoring, and batch record documentation. Under GMP, companies must maintain full traceability from raw material receipt to finished product distribution.
Good Laboratory Practice (GLP) ensures that non-clinical safety studies are conducted in a controlled and repeatable environment. It requires detailed protocols, accurate data capture, and a complete audit trail of all activities. This includes defined roles and responsibilities, approved standard operating procedures (SOPs), validated methods, and formal archiving practices.
Good Clinical Practice (GCP) applies to human clinical trials. It ensures that studies are ethically conducted and that data collected is credible. Requirements include subject consent tracking, investigator qualification, clinical site auditing, and real-time data monitoring. Electronic Trial Master File (eTMF) systems and audit-ready records are often part of GCP compliance efforts.
An effective audit management system plays a central role in supporting GxP compliance. It allows companies to:
- Schedule and document internal and external audits
- Track findings, assign responsibilities, and monitor follow-up actions
- Link audit results directly to CAPAs, SOP revisions, and training updates
- Maintain a secure, chronological audit trail
- Demonstrate regulatory readiness with real-time reporting and dashboards
Whether you’re preparing for an FDA inspection or a Notified Body audit, having these systems in place helps you move from reactive to proactive quality. Instead of scrambling to assemble documents, your team can respond with confidence and complete transparency.
QMS Standard: EU Annex 11
Annex 11 is part of the European Union’s GMP guidelines and focuses specifically on the use of computerized systems in regulated environments. It applies to any digital system used to support GMP activities, including manufacturing, laboratory testing, and quality management.
At its core, Annex 11 requires that computerized systems be validated to prove they consistently perform as intended. This means companies must document their system specifications, conduct validation protocols (IQ, OQ, PQ), and maintain clear records of testing and approvals.
Some of the key technical requirements include:
- System validation: Every computerized system used in a GMP context must be validated for its intended use. This includes functional testing, security assessments, and risk-based evaluations of system components.
- Data integrity: Systems must ensure that data is accurate, complete, and protected from unauthorized changes. This includes maintaining audit trails, using time-stamped entries, and ensuring that electronic records cannot be altered without proper authorization.
- Access control: Annex 11 requires controlled user access. Role-based permissions must be clearly defined and enforced through user accounts, secure logins, and electronic signature protocols.
- Change control: Any changes to the system, whether technical or procedural, must go through formal change control processes. This includes risk assessments, documentation updates, and testing before implementation.
- Backup and recovery: The system must have robust data backup procedures and a documented disaster recovery plan. These ensure data is not lost and that operations can resume quickly in the event of a system failure.
- Training and SOPs: All users must be trained on the system and the procedures that govern its use. Standard Operating Procedures must be in place for data entry, review, backup, recovery, and system maintenance.
For companies using an eQMS in Europe, compliance with Annex 11 is not optional. It is a critical part of maintaining GMP compliance. A well-configured audit management system and document control module can make it easier to meet these expectations, track validation efforts, and provide inspectors with the evidence they need during audits.
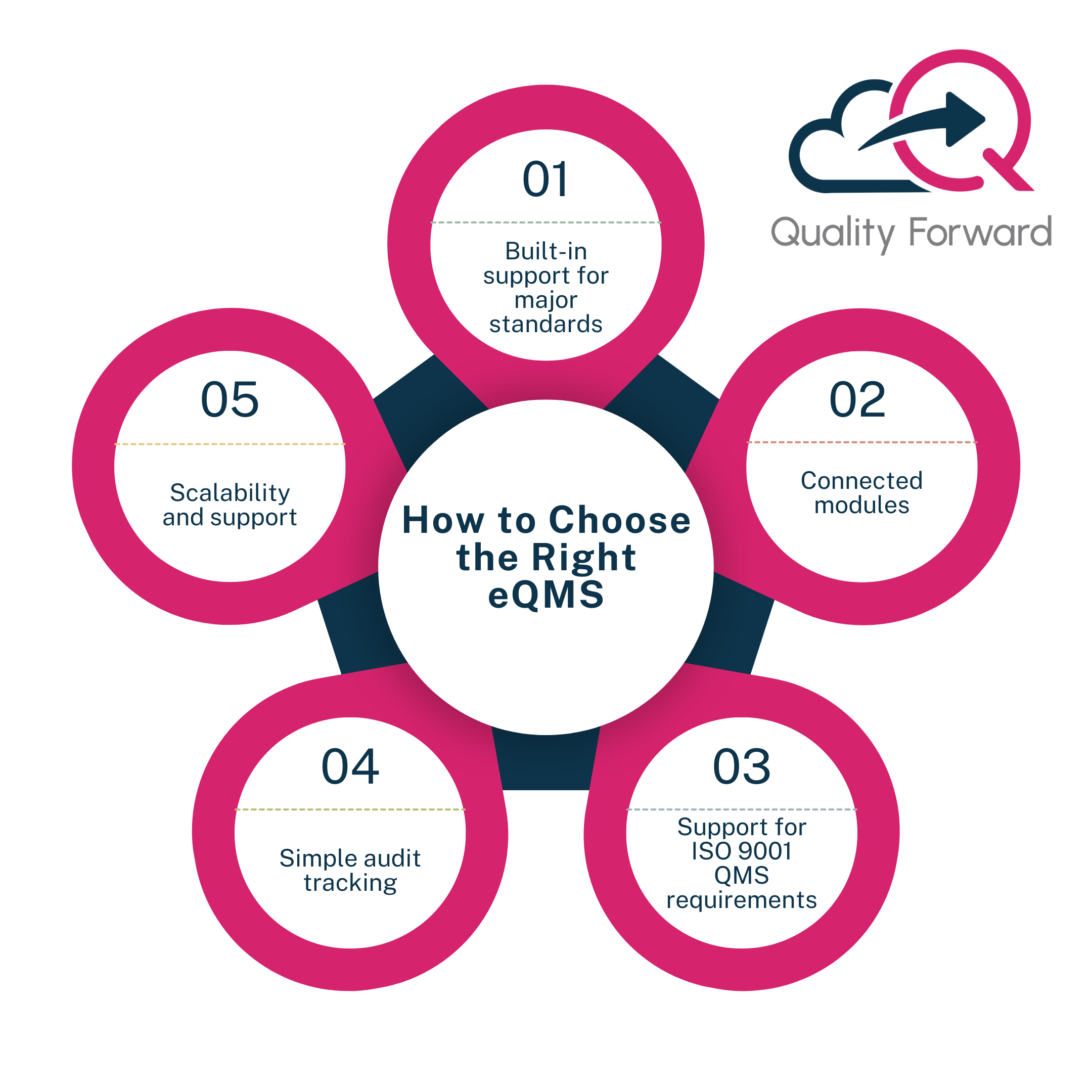
How to Choose the Right eQMS
When looking for a system that supports compliance with multiple QMS regulations, not all eQMS platforms are created equal. The best systems are not just designed for documentation. They are built to support the way regulated teams actually work.
Here are five things to look for:
Built-in support for major standards
Your system should align with ISO AND FDA QMS compliance out of the box. That means it should come with workflows and controls that support ISO 9001, ISO 13485, and 21 CFR Part 820.
Connected modules
Look for an eQMS that connects document control, CAPA management, audit tracking, and training in one system. This makes it easier to stay aligned across teams and reduces duplicate work.
Support for ISO 9001 QMS requirements
The system should help you meet the core elements of ISO 9001, including risk-based thinking, continuous improvement, and audit readiness. This makes ISO 9001 quality management more manageable and less time-consuming.
Simple audit tracking
Your platform should include a clear way to track audits from planning through follow-up. An audit management system should link findings to CAPAs and allow teams to collaborate on responses without relying on email threads or scattered spreadsheets.
Scalability and support
Your needs will grow. Pick a provider that understands the life sciences industry and offers reliable support, especially when regulations change or new requirements come into effect.
QMS Standards: Final Thoughts
Regulations are always evolving, but your approach to quality does not need to feel like you are starting from scratch each time. The goal is not to chase compliance but to create systems that hold up over time and support the way your people actually work.
QMS standards provide that structure, but how you apply them is what makes the difference. With the right tools in place, your quality system becomes less about reacting and more about staying ahead.
Yokogawa’s journey shows what that looks like in practice. By working with Quality Forward, they have been able to maintain control, adapt to new expectations, and build a system that fits the way their teams operate and not the other way around.
Compliance should not slow you down. With the right partner and platform, it becomes something you can rely on, not something you dread.
Frequently Asked Questions (FAQs): QMS Standards Compliance
Quality Management System (QMS) standards are frameworks that define how organizations document, control, and improve processes to meet regulatory and customer requirements. They ensure consistency, traceability, and continual improvement across operations.
Compliance demonstrates that products are designed, manufactured, and controlled in a way that meets safety and quality expectations. It helps companies reduce risk, streamline audits, and maintain trust with regulators and customers.
ISO 9001:2015 Quality Management provides the foundation for a process-based quality system. It emphasizes risk-based thinking, leadership involvement, and continuous improvement.
ISO 13485 is the QMS standard specific to medical devices. It builds on ISO 9001 by adding detailed requirements for design controls, risk management, supplier oversight, and traceability throughout the product lifecycle.
The FDA Quality System Regulation (21 CFR Part 820) sets mandatory QMS requirements for medical device manufacturers in the United States. It covers design, manufacturing, CAPA, complaint handling, and documentation.
Good Manufacturing Practice (GMP) defines minimum requirements to ensure products are consistently produced and controlled. GMP complements QMS standards by focusing on facility hygiene, process validation, data integrity, and quality control.
EU Annex 11 governs computerized systems used in GMP-regulated environments. It requires validation, access control, data integrity, and change management for all digital systems supporting GMP activities.
A modern eQMS integrates document control, training, CAPA, and audit modules. It helps teams comply with multiple frameworks (ISO, FDA, EU) in one system and reduces time spent on manual documentation and audit prep.


